Bakta Web
We provide a dedicated Bakta web version available via https://bakta.computational.bio.
Overview
The Bakta mainpage contains multiple sections for the user input. A textfield to paste your fasta sequence as well as a file input to upload your sequence as a Fasta file.
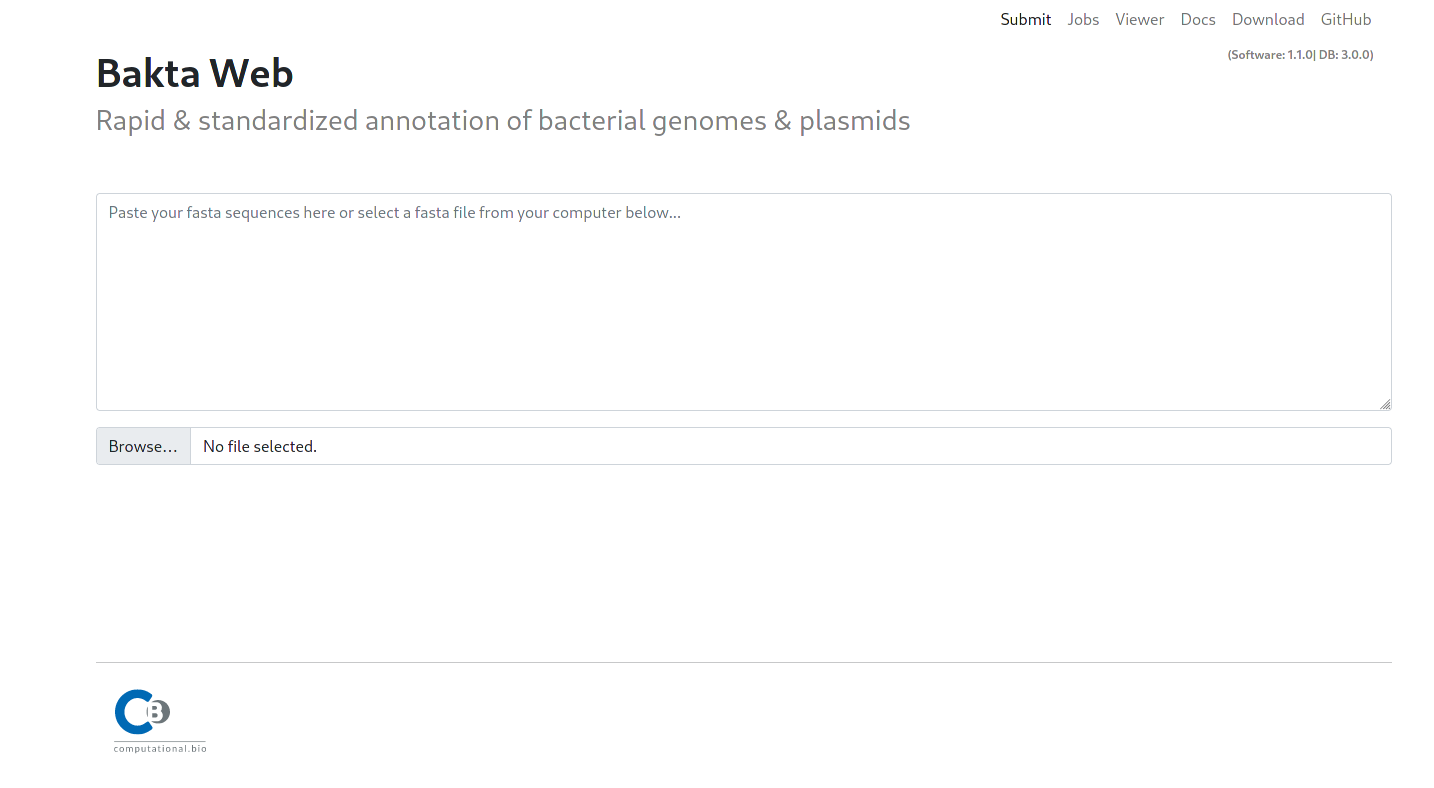
The menu contains multiple subsections:
Submit: The mainpage for submitting new Jobs
Jobs: Overview of all submitted Jobs and their status
Viewer: Visualizing genome annotation results conducted with the local CLI version
Docs: documentation
Download: Download the Bakta tool and database
GitHub: Github repository.
Submit options
After choosing a sequence, users can set additional options before submitting.
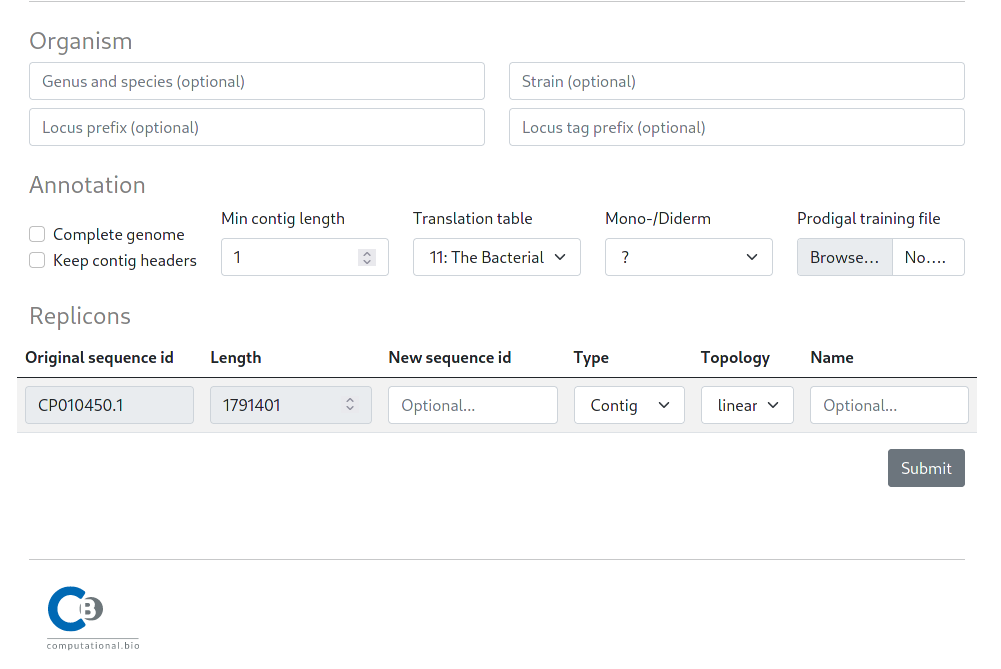
The submit options are split in three sections. An Organism section that allows users to specify additional (optional) description tags for the submitted fasta sequence, an Annotation section to specify the annotation settings and a Replicons section to provide optional sequence metadata, e.g. completeness and topology.
Monitoring Jobs
Submitted jobs are monitored automatically in the Jobs tab.
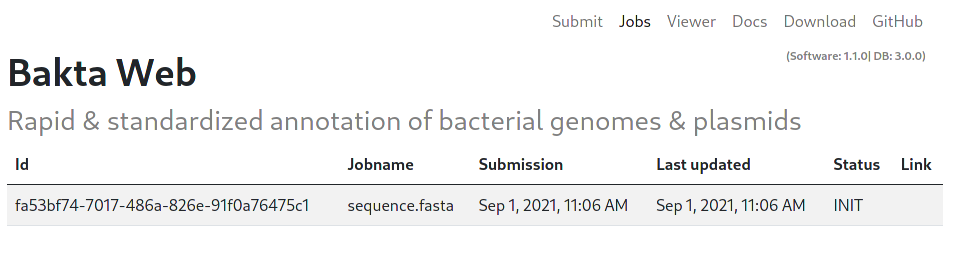
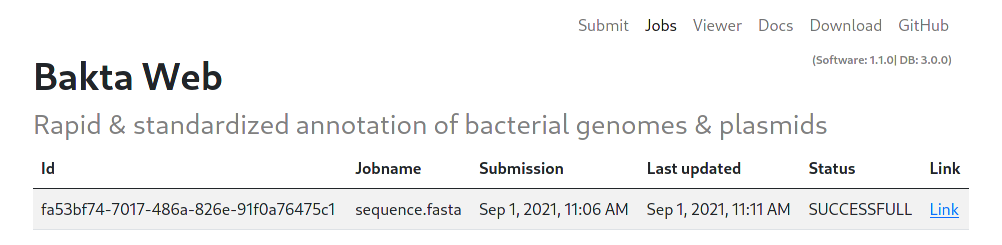
All jobs start with the INIT status. This indicates an initializing status, as well as a waiting position in the queue. A running Job is indicated by the RUNNING status.
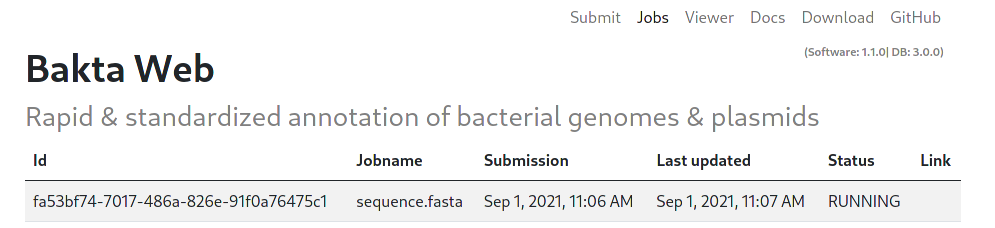
Finished jobs have the SUCCESFULL status and include a LINK to see the results in the Viewer tab.
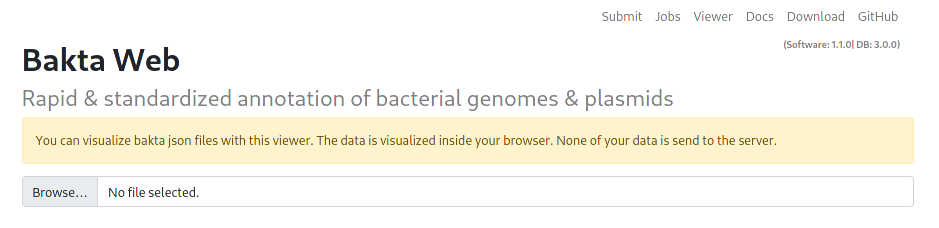
Visualization
Results can be visualized via the Viewer tab, to visualize local files users can choose a local Json file for visualization. This visualization happens entirely and exclusively in the Browser, no data is uploaded to the server.
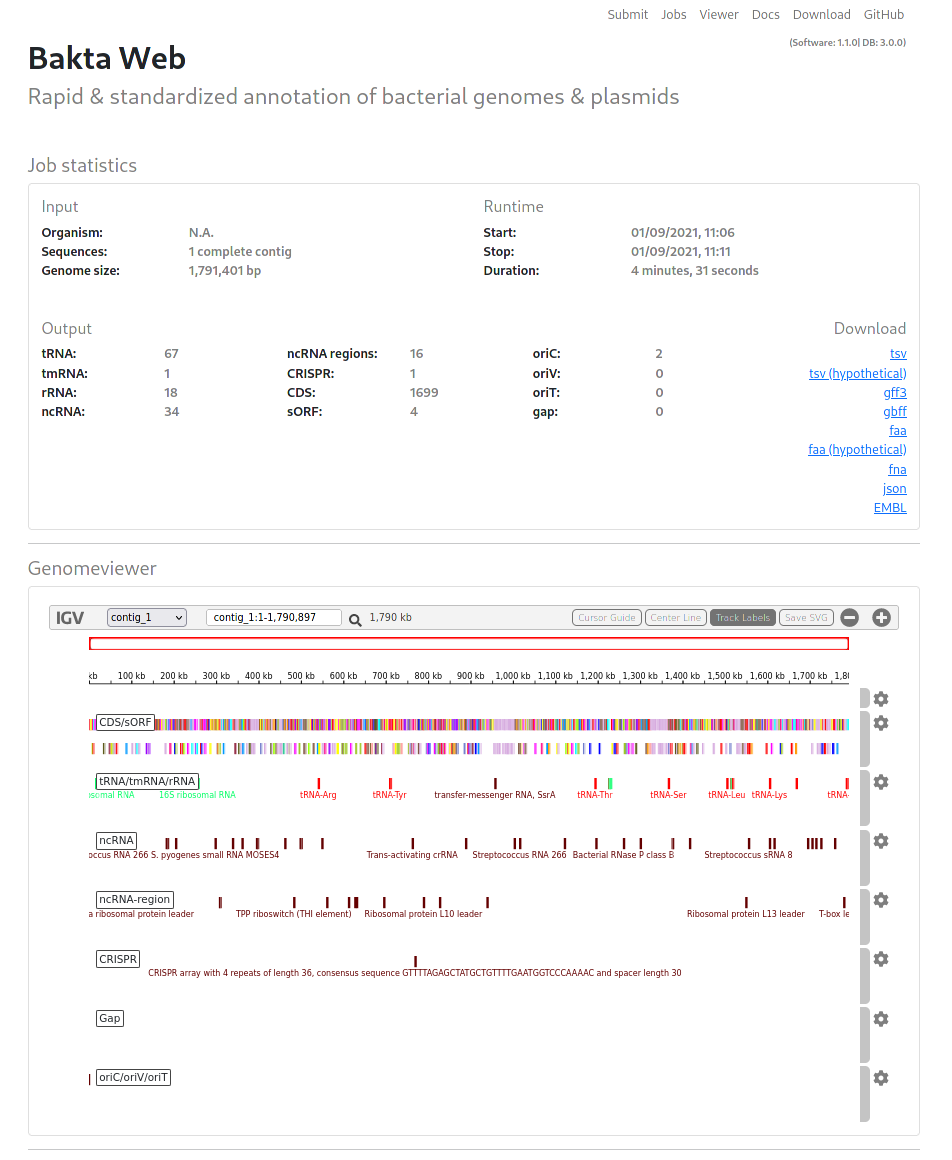
Results
Visualized results contain three sections:
Job statistics: Contains a general overview of the annotated genome.
Genomeviewer: An IGV-based genome browser to visualize annotated features.
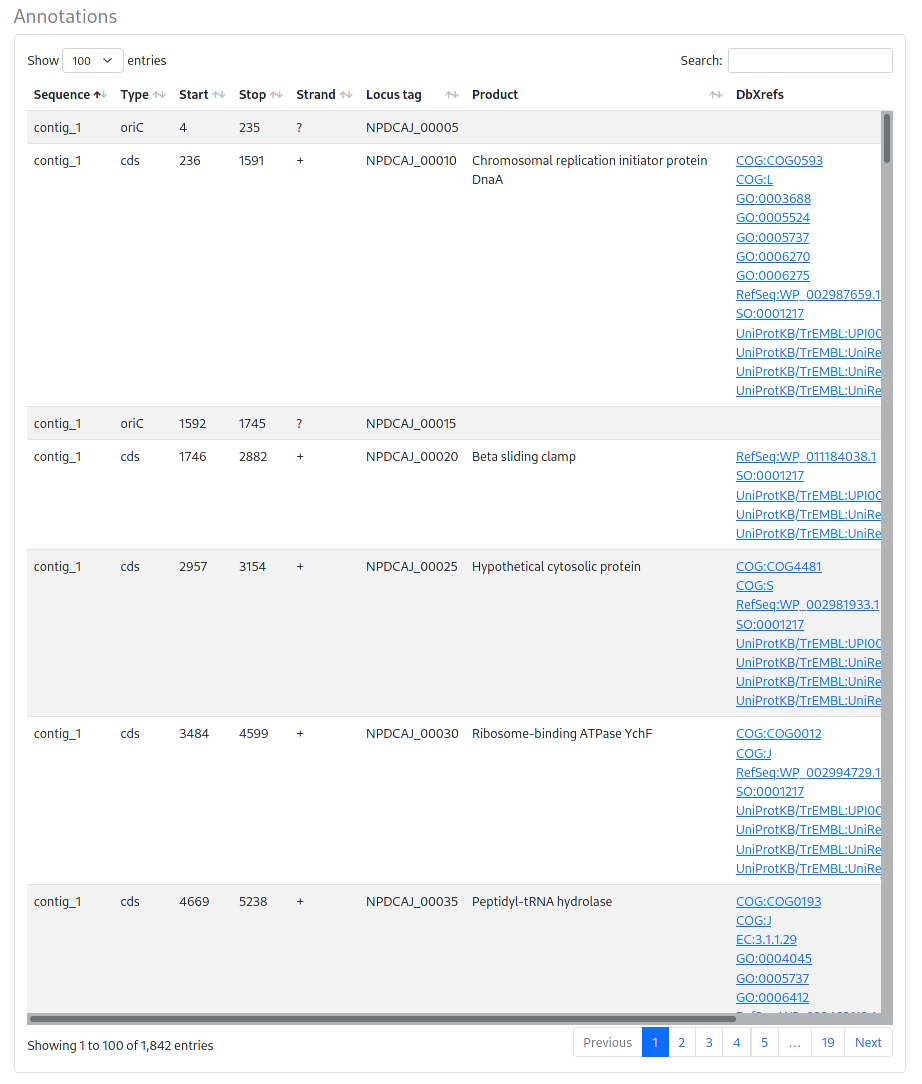
Annotations: A comprehensive list of all annotated features, including DB cross references to multiple common databases.